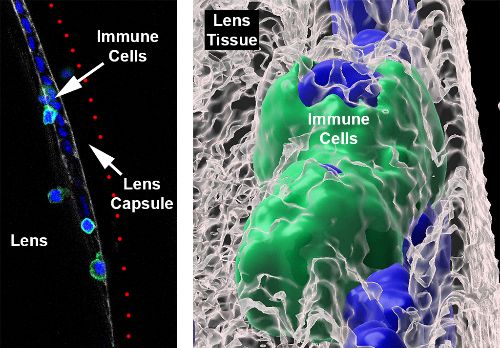
Immune Cell Presence on the Crystalline Lens in Uveitis
Despite being susceptible to a range of potential threats—whether
they come from pathogens, chemicals, radiation, or general oxidative
damage—it is surprising that ocular tissues such as the cornea,
crystalline lens, and retina are thought to be immune privileged, that
is, lacking immune cells. So how do these critical tissues protect
themselves? Closer investigation by researchers in previous work
revealed, for example, that mice engineered to lack a key protein for
crystalline lens formation showed immune cells trying to fix the resulting malformed lens. Other researchers observed immune cells in the cornea after damage to the lens, and vice versa immune cells arriving at the lens surface, acting like sentinels to protect this tissue after damage to the cornea (the external ocular surface). In a new study,
researchers at Thomas Jefferson University show that immune cells
respond to the lens not only after acute eye injury but also in chronic
inflammation. Using high-resolution microscopy and a mouse model of
uveitis, or inflammation to the middle layer of the eye, the researchers
studied the role of immune cells in relation to cataracts associated
with uveitis.
Unlike
the sentinels seen after ocular surface injury, the researchers report,
"In this case, it was like a battering ram. There were dozens of immune
cells, and different types of them, including T-cells and macrophages.
It’s clearly a robust immune response and could reflect in part that
inflammation in uveitis is so severe." Similarly, it was previously
thought that the lens capsule surrounding the lens protects it from the
increase in immune cells that populate the aqueous and vitreous chambers
during active uveitis. However, high-resolution confocal z-stacks
and scanning electron microscopy revealed that immune cells were
actually integrated into the lens capsule, increasing not only in number
but also in depth (invasion) as the uveitis progressed. Moreover, the
immune cells were observed to be able to penetrate the thick lens capsule,
infiltrate into and embedding themselves in the lens tissue. Although
most of the cells were gone as the uveitis began to resolve, some of
these immune cells remained integrated into the lens capsule and lens
tissue. Underscoring the complexity of the presence of immune cells in
the eye, a topic that has thus far been understudied, the senior author
of the study explains, "Till now, the mechanisms for damage that happen
in this region of the
eye after uveitis have been poorly understood. For the
first time, we’ve been able to provide evidence that immune cells could
be driving this damage, particularly to the lens.”
{kind=link}
SINE RNA Receptor DDX17 Mediates Lupus & AMD
Jayakrishna Ambati, MD, has been a prolific researcher, publishing three
papers this year regarding his discoveries connecting atrophic macular
degeneration (AMD) with cytoplasmic cDNA and its activation of the
inflammasome. His fourth paper this year presents another finding
linking triggers of the inflammasome with another disease: lupus. Ambati
remarks, "It appears that the new inflammatory pathway we identified
could be therapeutically targeted for many chronic diseases." The
inflammasome, or pairs of inflammasomes, responsible in this case is the
NLRC4-NLRP3 inflammasome. Each are individually large multi-protein
complexes that play an important role in protecting the body from
pathogens. However, in noninfectious, chronic inflammatory diseases such
as lupus and macular degeneration, the researchers found that the NLRC4
inflammasome includes the NLRP3 inflammasome, and is instead
independent of NLR family apoptosis inhibitory proteins (NAIPs) that are
classically
required for NLRC4 inflammasome assembly after bacterial infection. In
the "sterile" environment of chronic diseases, that is, in the absence
of pathogenic etiologies, researchers wondered what triggers the
formation of inflammasomes.
The new research uncovered that the NLRC4-NLRP3 inflammasome is
triggered by short interspersed nuclear element RNAs (SINE RNAs), mobile
genetic elements known as retrotransposons that make up more than 10%
of our genome and is transcribed in response to cellular stresses. SINE
RNAs were found to be elevated in both lupus and AMD, among other
diseases in previous work. Notably, using human cells transfected with
SINE RNA, the research team identified a receptor called DEAD-box
helicase 17 (DDX17). When DDX17 was unavailable, there was less
interaction between the NLRC4 and NLRP3 proteins in SINE RNA-transfected
cells, suggesting that DDX17 is an essential mediator in sterile
activation of the NLRC4 inflammasome by SINE RNAs. The finding that
DDX17 is colocalized with SINE RNA in the cytoplasm is confirmed in
white blood cells isolated from people with active lupus. Additionally,
subretinal injection of SINE RNAs in a mouse model of atrophic macular
degeneration led to retinal pigmented epithelium (RPE) degeneration
in wildtype mice but not in mice engineered with genetic loss of Nlrc4 or Ddx17,
demonstrating that the NLRC4 inflammasome and its receptor DDX17 are
both necessary to trigger the retinal disease. The finding that there
are two inflammasomes involved, both of which are necessary to form an
active NLRC4 inflammasome when the complex is triggered by SINE RNAs,
also informs therapeutic strategies for both the NLRC4 and NLRP3
inflammasomes. Ambati further comments, “[N]ow that we know what is the
sensor—at least a sensor—of SINE
retrotransposons, that opens up a whole new intersection between RNA
biology and immunology.”
Metformin Explored as a Treatment for L-ORD
Late-onset retinal degeneration (L-ORD) is a rare genetic disorder of
autosomal dominant inheritance. Specifically, L-ORD is caused by a
missense substitution in the gene that encodes the protein CTRP5, leading to choroidal neovascularization
and deposits of apolipoprotein E (which is involved in lipid metabolism
within the retina), and retinal pigmented epithelium (RPE) atrophy (which
contain an abundance of fatty acids and lipids). Symptoms begin with
nyctalopia, progressing to central vision loss by the sixth decade.
Scientists at the National Eye Institute are studying the disease by
developing a "disease-in-a-dish" model from induced
pluripotent stem cells (iPSCs) to make RPE cells from skin fibroblasts;
the fibroblast samples were collected from two siblings with L-ORD (L-ORD-iRPE) and compared with two unaffected siblings who lacked the disease-causing mutation. The L-ORD-iRPE were observed to be dysmorphic (deformed), also showing deposits
of apolipoprotein E near the tissue and abnormal secretions of vascular
endothelial growth factor (VEGF); these cells also had reduced
secretions of both normal and mutant CTRP5 proteins.
Computer modeling of the proteins showed that mutant CTRP5 was less likely to bind with cell receptors that regulate lipid metabolism, in turn leading to chronic activation of AMP-activated protein kinase (AMPK), a key regulator of energy homeostasis and lipid metabolism, such as of apolipoprotein E. One of the researchers explains, "AMPK in the RPE itself is a key regulator of the conversion of DHA [docosahexaenoic acid] into protective mediators [against oxidative damage and mutations]." Chemically inhibited AMPK in
the L-ORD-iRPE cells led to fewer apolipoprotein E deposits and less abnormal secretion of VEGF. Finally, the researchers tested two therapies on the L-ORD-iRPE cells: a gene augmentation of normal CTRP5, and modulation of AMPK with anti-diabetes drug metformin. Both strategies prevented signs of L-ORD in the RPE model. Senior author of the study states, "Importantly,
we now have two potential strategies to
disrupt the L-ORD disease process. While gene therapy may be years away,
metformin is a drug that’s long been used to treat diabetes." Although
L-ORD is a rare genetic disease, it shares some characteristics with the
much more common age-related macular degeneration (AMD). The
researchers hope that studying this model, and the effect of metformin,
will benefit diseases caused by RPE changes.
Association Study Shows Link between AMD Genetic Risk Factors and Thinner Retinal Layers
Age-related macular degeneration (AMD) is a leading cause visual
impairment and loss in Western countries, and especially among
individuals 55 years of age and older. Researchers in the U.K. looked at
medical records from the U.K. Biobank, which includes retinal scans and
genetic data from over 30,000 patients. In particular, they compared
the latest data of 34 known genetic risk factors of AMD (which together
comprise 46% of the disease's genetic variance) with macular thickness,
as measured with spectral-domain optical coherence tomography (SD-OCT).
The authors report, "Our analysis has interestingly shown that, in the
presence of high
genetic risk for AMD, there is a significant decrease in the thickness
of both the ISOS-RPE and the RPE-BM which may suggest that premature RPE
thinning could be a major contributory factor." The inner-segment outer
segment-retinal pigment epithelium (ISOS-RPE)
thickness measurement, is of particular interest in that it represents
the photoreceptor outer segments where light transduction takes place.
The researchers conclude, "Our study highlights the premorbid influence
of AMD genetic risk
variants on macular thickness and may provide mechanistic insight into
the pathophysiology of this debilitating disease." Because these
structural changes occur prior to onset of disease symptoms or overt
clinical signs, they could provide an early assessment of disease risk
to guide healthy habits.
Regulators of Adult Visual Cortex Plasticity in Mice
Learning and recovery from injuries depend on the brain's plasticity.
This plasticity between neuronal connections relies heavily on the
network of macromolecules in between and surrounding the nerve cells,
known as the extracellular matrix (ECM). As the brain becomes more
mature, the stability of the ECM increases, providing a scaffold for the
existing arrangements and synaptic circuits of nerve cells. New
experiences require that this extracellular matrix be loosened in order
for new connections to form. Similarly, when the brain experiences an
injury such as a stroke, it needs to reorganize itself and form new
connections. This balance between stability and plasticity is regulated
by the proteolytic activity of enzymes such as matrix metalloproteinases
(MMPs), which "digest" the ECM in order to "loosen" it. Researchers in
Germany studying the visual cortex of mice showed that blocking the
matrix metalloproteinases MMP2 and MMP9 can have opposing effects
depending on whether the brain is sick, such as after stroke, or
healthy. In the primary visual cortex of healthy mice, blocking MMP2 and
MMP9 led to decreased ocular dominance plasticity. In mice studied
immediately after a stroke, inhibition of MMP2 and MMP9 (which spike for
a short time after a stroke) rescued neuronal plasticity that had been
compromised by the stroke, that is, the MMPs had a therapeutic effect.
The researchers point out that the intentional inhibition of the
metaloproteinases immediately after inducing an experimental stroke was
in order to simulate treatment. The authors argue that these findings
show that levels of MMPs must be precise and optimal in the brain, as
both too low or too high an amount can prevent neuronal plasticity.
In Other News
(1) Vision's effect on the development of hearing investigated
(2) Candy-like models to make STEM accessible to students with visual impairment
(3) Visual behavior and autism prediction in infants
No comments:
Post a Comment